

Since Einstein’s analysis of Brownian motion in 1905, scientific world has accepted that matter around us is made out of tiny particles called atoms. These atoms themselves consist of a small nucleus made up of protons and neutrons, surrounded by a “cloud” of electrons. Behavior of electrons and nuclei is very well described by combining quantum mechanics and electromagnetic
theory. This combined theory can be used to predict optical, thermal, mechanical, and electrical properties of matter.
In my master’s thesis I studied the electron density in crystalline matter using computational methods. In particular, I focused on combining the quantum-mechanical behavior of both nuclei and electrons. Experimentally the electron density of crystals can be measured, for example, using X-ray crystallograpy. Taking into account the quantum nature of nuclei is required to theoretically or computationally match the experimentally observable density.
In quantum mechanics, a system of electrons and nuclei is described by a Schrödinger equation where the potential function is the electrostatic (Coulombic) potential. This equation is famously difficult to solve, and closed-form solutions exist only for the simplest of cases, such as the hydrogen atom (2 particles). More complex systems, such as the Helium atom (3 particles), require the use of approximations. The theoretical part of my thesis lays out a sequence of approximations, which together allow the electron density of crystals to be found computationally. A very common starting point, which I also used, is the Born–Oppenheimer (BO) approximation. This approximation assumes electrons to instantaneously adapt to “movements” of relatively much heavier nuclei, allowing the nuclear and electronic parts of the Schrödinger equation to be solved in sequence, instead of trying to solve for all particles at once. Further approximations I used are the harmonic approximation for nuclear dynamics, the use of small repeated supercells (64 atoms) to represent infinite crystal lattices, and the use of approximate functionals (PBE) in density functional theory.
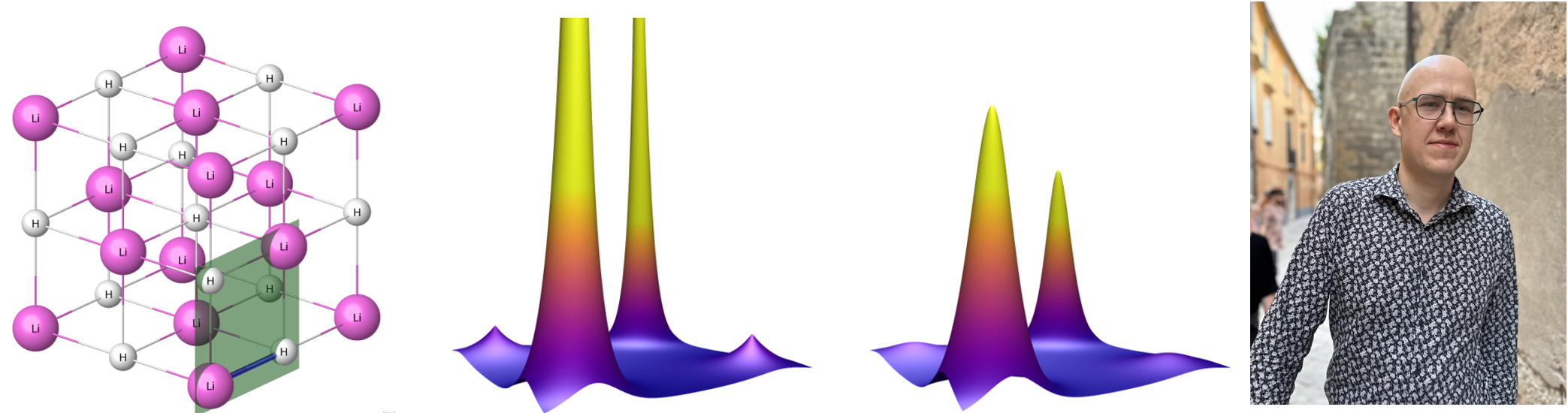
My thesis makes a distinction between two types of electron density: static electron density, where nuclei are treated as stationary particles, and dynamic electron density, where nuclei are modeled quantum mechanically. Examples of computed densities in lithium hydride (LiH) are seen in Figure 1. This figure neatly summarizes the qualitative effect of the quantum nature of nuclei: electron density peaks are lowered to produce a smoother electron density. In crystallography, this behavior is known as thermal smearing1. In addition to LiH, which is interesting due to having light nuclei, I studied a more commonly known crystal: diamond.
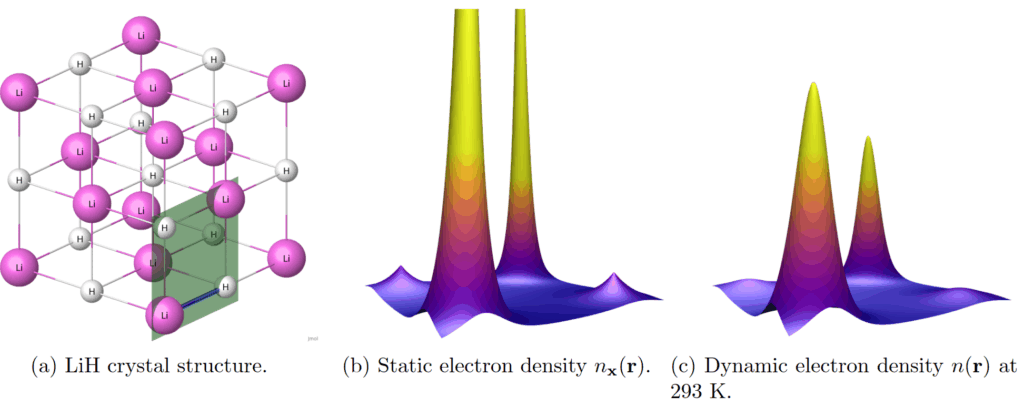
I computed static electron densities using computational DFT suite Quantum ESPRESSO. This suite was also used to find interatomic force constants (harmonic), from which distributions of nuclear configurations were computed. I implemented three different methods for computing the dynamic electron density by combining the distribution of nuclear configurations with computed static electron densities. Theoretically the most accurate of these was the Monte Carlo method, where an average is computed over static electron densities obtained from a sample of nuclear configurations. The Monte Carlo method was also the most computationally expensive method. The cheapest method I used is based on the convolution approximation, which assumes that an electron cloud surrounding a nucleus moves around with the nucleus with no change in shape. Under this assumption dynamic electron densities can be computed as convolutions using fast Fourier Transforms (FFT).
The convolution approximation method was found to closely match the more accurate Monte Carlo method, with largest relative difference being 0.6% in diamond and 2.2% in LiH. These were confirmed to be real errors resulting from the convolution approximation, not simply caused by the inherent uncertainty of the Monte Carlo method. This work was the first computational validation of the convolution approximation in crystals.
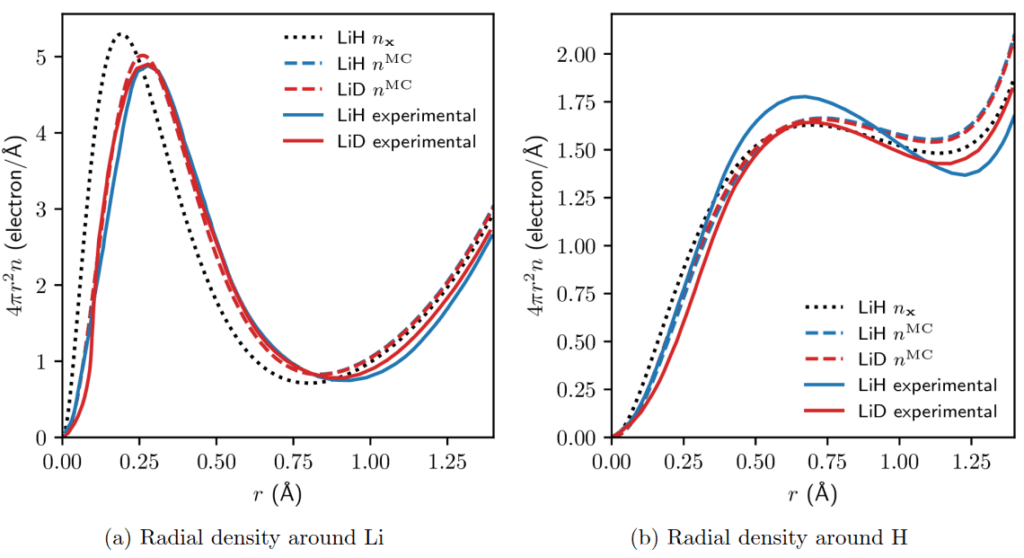
I compared the computed densities with experimental observations, demonstrating the importance of taking the quantum nature of nuclei into account. This can be seen in Figure 2, which shows my computational radial electron densities compared with prior experimental results. The experiments performed by G. Vidal-Valat et. al. on lithium hydride (LiH) and lithium deuteride (LiD) showed an anomaly that can be seen in Subfigure 2b: there is a significant deviation in electron density depending on the hydrogen isotope. This difference was suggested to be due to a breakdown in the Born–Oppenheimer approximation. My computational work shows that when making the Born–Oppenheimer approximation, this difference is expected to be rather small. A question for future research is thus raised: can the previous experimental results be replicated using new computational methods that don’t make the Born–Oppenheimer approximation?
The full thesis can be found here: Effect of nuclear quantum nature on electron density in crystalline solids.
1This term is somewhat inaccurate, since such smearing would also happen at 0 K due to zero point oscillations.



{kind=link}
{kind=link}
{kind=link}